

A gene is a coded genetic material called DNA. Each gene contains information required to produce gene products, mainly proteins, which are needed for cell function and for organisms to survive. Just as in our daily life we buy or use things, as we require, similarly these genes make gene products in the body as when required. This on and off mechanism is called Gene Expression. Gene expression is the process by which the information encoded in a gene is used to direct the assembly of a protein molecule (Gene expression).
Regulation of Gene Expression:
Genes are expressed either as RNA or proteins. However, as mentioned earlier not all gene products are required at all point of time in a cell and also the amount of protein required might vary as per the demand of the particular organ. So, based on the external and internal requirements and other environmental factors, the cells need to decide how much gene expression is necessary for it.
The amounts and types of mRNA molecules in a cell reflect the function of that cell. Thus, the primary control point for gene expression is usually at the very beginning of the protein production process — the initiation of transcription. RNA transcription makes an efficient control point because many proteins can be made from a single mRNA molecule. Eukaryotic transcripts are also more complex than prokaryotic transcripts. Different cell types have varying gene expression profiles because of presence of distinct transcription regulators. DNA sequence usually has a promoter sequence to which RNA polymerase can bind and thus start the transcription process. Along with the promoter sequences, there are also enhancer sequences on DNA, which provide binding sites for regulatory proteins that affect RNA polymerase activity. These regulatory proteins can either increase or decrease the transcription process, thus influencing the expression of a gene in a cell (http://www.nature.com/scitable/topicpage/gene-expression-14121669),(http://study.com/academy/lesson/what-is-gene-expression-regulation-analysis-definition.html)
Epigenetics and DNA Methylation:
One important aspect in gene expression is that it does not only depend on the gene’s DNA sequence but is also influenced by the epigenetic or external environmental factors. Thus, Epigenetics is the study of changes in gene expression, which is controlled by external factors other than the gene’s DNA sequence, and these changes are heritable in nature. Epigenetic changes can switch genes on or off and determine which proteins are transcribed. It is also involved in many cellular processes and thus all our cells have the same DNA but get differentiated into different types of cells in the body such as neurons, liver cells, pancreatic cells, etc. (http://www.nature.com/scitable/topicpage/epigenetic-influences-and-disease-895)
Source:journal.frontiersin.org
DNA methylation is an epigenetic mechanism in which a methyl (CH3) group is added to a DNA. It usually happens in a region where a cytosine nucleotide is located next to a guanine nucleotide linked by a phosphate, this is a called a CpG site. In the bulk of genomic DNA, most CpG sites are heavily methylated while CpG islands (sites of CpG clusters) in germ-line tissues and located near promoters of normal somatic cells, remain unmethylated, thus allowing gene expression to occur. When a CpG island in the promoter region of a gene is methylated, expression of the gene is repressed (it is turned off). ( http://www.whatisepigenetics.com/dna-methylation/)
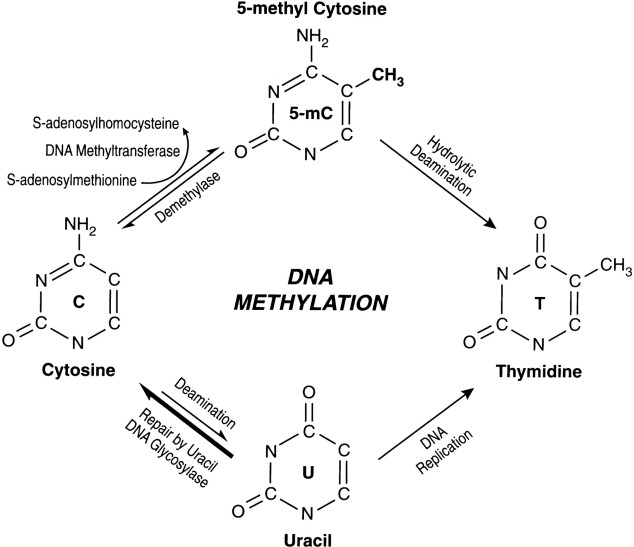
Source: www.bloodjournal.org
In previous blogs we have discussed mainly about the CYP2C19 gene, but for this part of the blog we will be analyzing gene expression for CYP2C9 gene. Using the Tissue-specific Gene Expression and Regulation (TiGER) database curated by John Hopkins University, we can observe the tissues in which the CYP2C9 is mainly expressed:
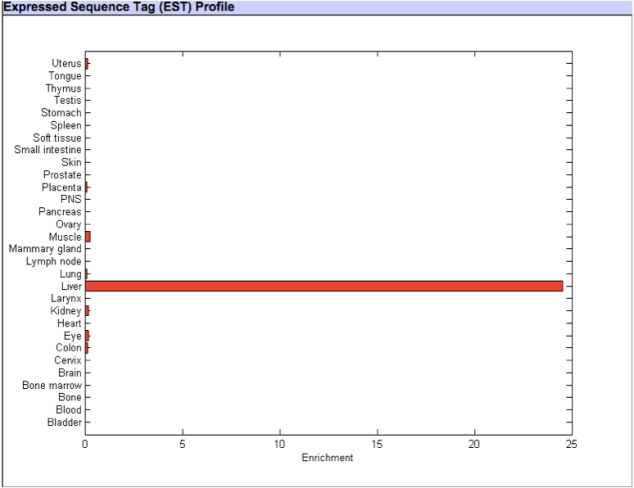
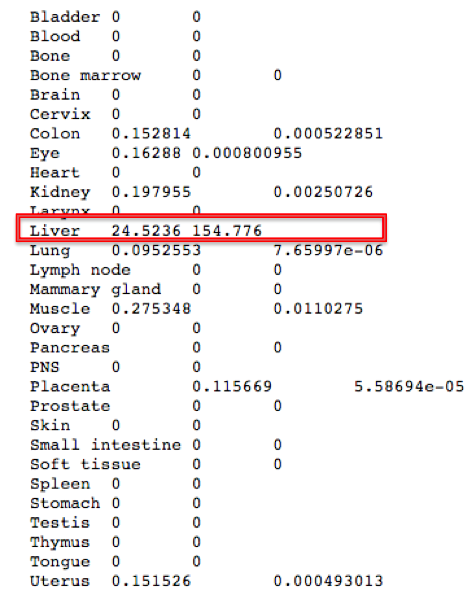
CYP2C9 Gene expression – TiGER database
From both figures above of the Expressed Sequence Tag (EST) Profile, it can be observed that the CYp2C9 gene is mainly expressed in the liver with a small amount expressed in uterus,muscle,colon,kidney and eye. (http://bioinfo.wilmer.jhu.edu/tiger/db_gene/CYP2C9-index.html).This information is concurrent with the genotype and phenotype details of CYP2C9 gene mentioned in OMIM.org . As per OMIM, the CYP2C9 gene belongs to the cytochrome P450 enzyme family which are mainly responsible for metabolism of drugs such as anti-coagulant warfarin, anti-diabetic drugs tolbutamide and glipizide and anti-convulsant phenytoin. These drugs mostly have hepatic metabolism pathway and thus, we could agree with the TiGER database of the CYp2C9 gene being specifically expressed in liver.
Similar results were seen with The Human Protein Atlas database as seen below:
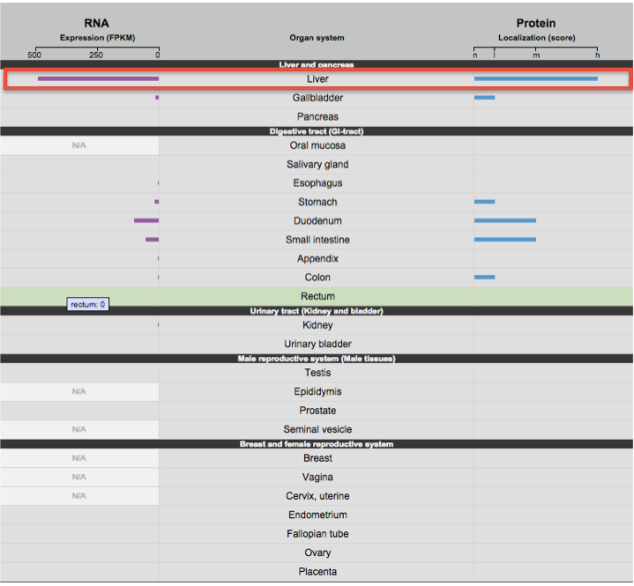
The RNA Expression and Protein Localization for the CYp2C9 gene is highest in liver followed by duodenum, small intestine, colon,kidney and appendix.
In order to study the epigenetic effect through DNA methylation in the CYP2c9 gene, the NCBI Epigenomics ‘Browse Experiments’Tool was used to yield the following results:
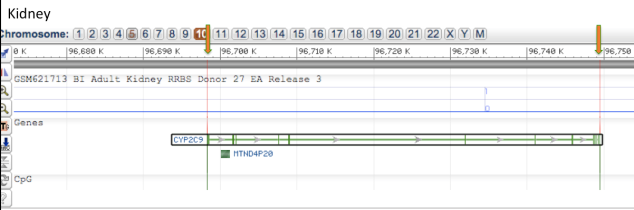
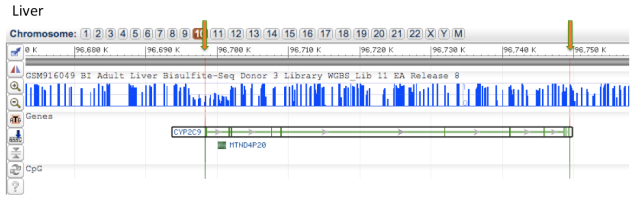
Epigenomic Analysis of DNA methylation of Kidney and Liver tissue for CYP2C9 gene
Comparing the DNA methylation of CYP2C9 gene on chromosome 10 in adult kidney and adult liver tissue shows that there are no CpG islands within the gene which is highlighted by the orange arrows. Also it can be seen that the kidney tissue has almost no methylation, whereas the liver tissue has very high methylation across the gene.
Lastly, to compare RNASeq and Microarray technologies for evaluation of differential gene expression of CYP2C9 gene, I used the huge Neuroblastoma dataset and narrowed down my search to the CYP2C9 gene in the gene data bank. I found only one entry for the gene, which is documented below:

As seen in the figure above, the differential score for Tumor Stage 1 and Tumor Stage 4 has not been reported. But for Tumor Stage 4S the differential score was 50.4 for RNASeq which down regulated to 38 for the Microarray technology. Thus, it proves that RNASeq is a better differential technique to use as compared to Microarray.


























