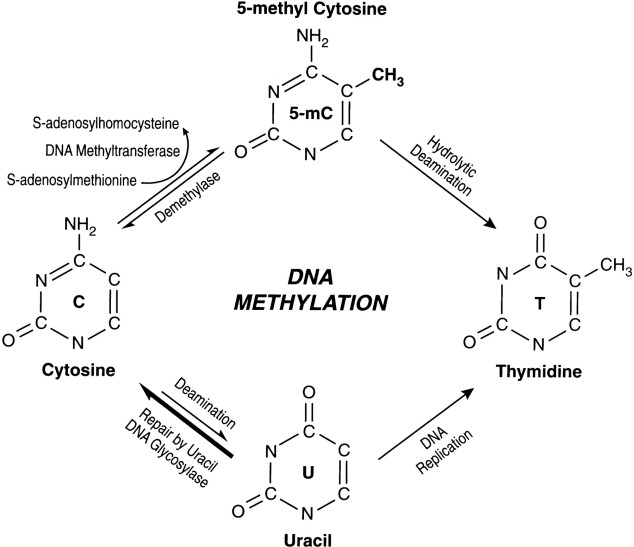
Living systems are complex in nature and so it becomes difficult to predict their functioning or behavior by just studying an individual part of it. Thus, Systems Biology involves the study of systems of biological components, which may be molecules, cells, organisms or entire species (Systems Biology ). It is a combination of various study disciplines including biology, computer science, engineering, bioinformatics, physics and others. This integrative approach helps scientists to design predictive models of such complex systems, which further aid in better understanding of how these systems respond to changes in time and environment. Based on the results of these predictions, target drugs and precision treatment approaches can be designed,new biomarkers for disease can be discovered as well as patients can be stratified based on their genetic profiles. On biological level, our bodies work on the principle of ‘Network of Networks’. Any living body is made up of many networks, that communicate with each other based on internal and external changes to effectively perform their biological function. These networks can be at cellular level, molecular level, genomic level,etc.Systems biology looks at these networks across scales to integrate behaviors at different levels, to formulate hypotheses for biological function and to provide spatial and temporal insights into dynamical biological changes .(https://www.systemsbiology.org/about/what-is-systems-biology/ )
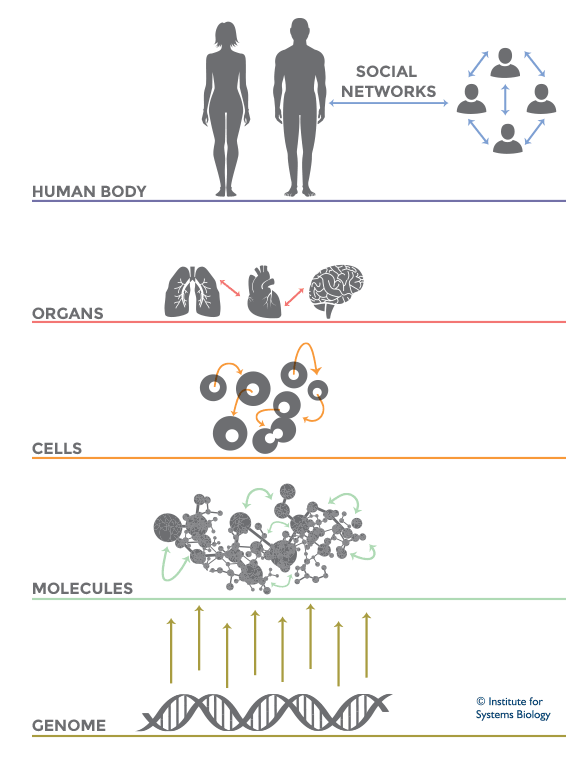
Network of Networks theory in Systems Biology (Source: Institute for Systems Biology)
Biological Pathway:
The cells in various organs of our body are constantly interacting and receiving chemical cues from external and internal environment in the form of injury, infection,need for food, etc.In order to react to these cues and produce certain products such as fat,protein,etc o to perform a required cell function so that the develops and stays healthy, these cells send and receive signals through biological pathways. Thus, a biological pathway is a series of actions among molecules in a cell that leads to a certain product or a change in the cell. Such a pathway can trigger the assembly of new molecules, such as a fat or protein. Pathways can also turn genes on and off, or spur a cell to move.(Biological Pathway).
Various resources were used to study the various molecular interactions of CYP2C19 gene and its involvement in biological system.
The first database that we searched was Geneontology.org (GO). This is an open access database and upon searching for Ontology information for the CYP2C19 and CYP2C9 gene, not enough information was found to be registered in there.So just to give an idea to the readers of this post about how this source is useful for pathway analysis, CYP2D6 gene was considered for the search query, which gave the following result:
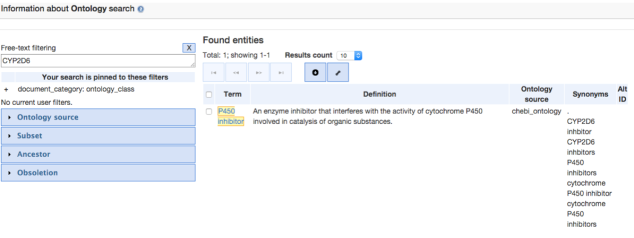
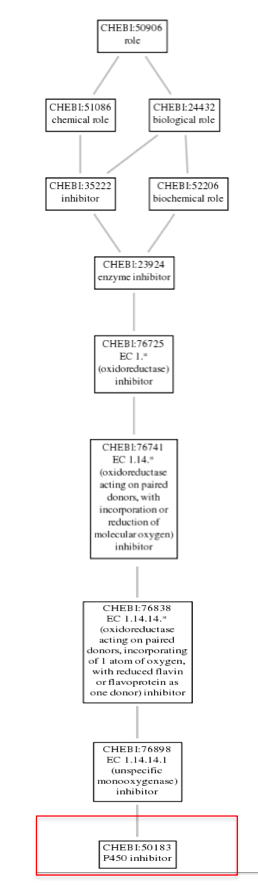
Further, navigating through the P450 inhibitor option gave us some Term information, that included the GO Accession ID :CHEBI:50183 for the CYP2D6 gene. Clicking on the Graph view, gives the ontology of CYP2D6 as shown in the image below:
KEGG: Kyoto Encyclopedia of Genes and Genomes :
KEGG is an open source database for understanding the various functions, interactions and pathways involving cells, molecules, organism and ecosystem of a biological system. On entering our gene name in the KEGG Pathway search option, around six different pathways involving the CYP2C19 gene are obtained. I just selected the most relevant Drug Metabolism- Cytochrome P450, which is in line with the information we have discussed in previous blogs that the major role of the CYP2C19 gene is in the metabolism of drugs such as mephenytoin, proguanil,warfarin,etc.
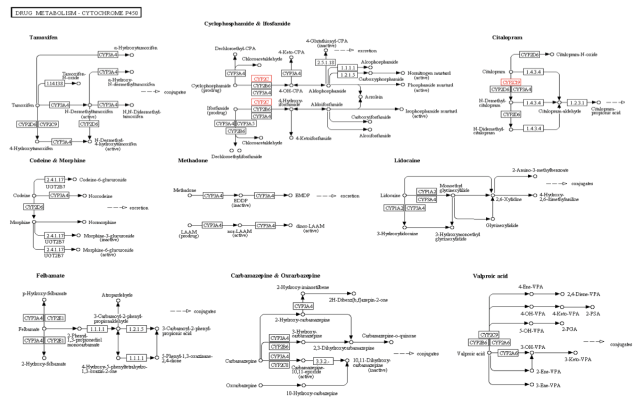
Kegg Pathway Text Search for the CYP2C19 Gene: Drug Metabolism – Cytochrome P450 Pathway ( Entry-map00982)
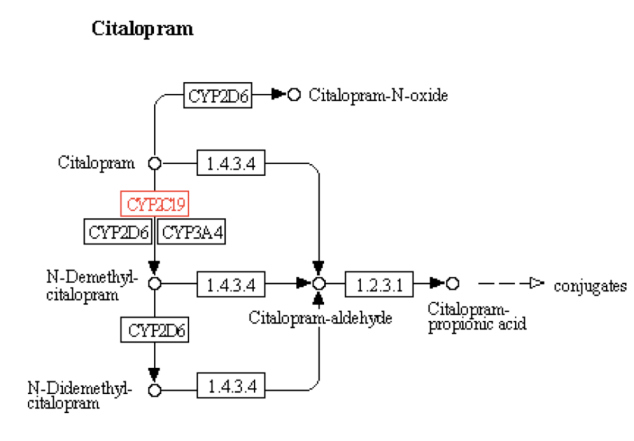
The particular metabolism pathway were the involvement of the CYP2C19 gene is highlighted in above figure is for the drug Citalopram which is as follows:
Ingenuity Pathway Analysis (IPA) database is a highly curated source which does not have an open access. It is a web based software application, that uses powerful algorithms to identify regulators, relationships, mechanisms, functions, and pathways relevant to changes observed in an analyzed dataset.
Initial search for the CYP2C19 gene in the Genes and Chemicals tool of the IPA yields the following result, which shows that none of the drugs are associated with this gene as highlighted with a red box in the below figure:

On selecting the CYP2C19 gene from the above entry and adding it to my new pathway and building it with the help of Grow tool where number of molecules were limited to 25 gives the following pathway in Organic view:
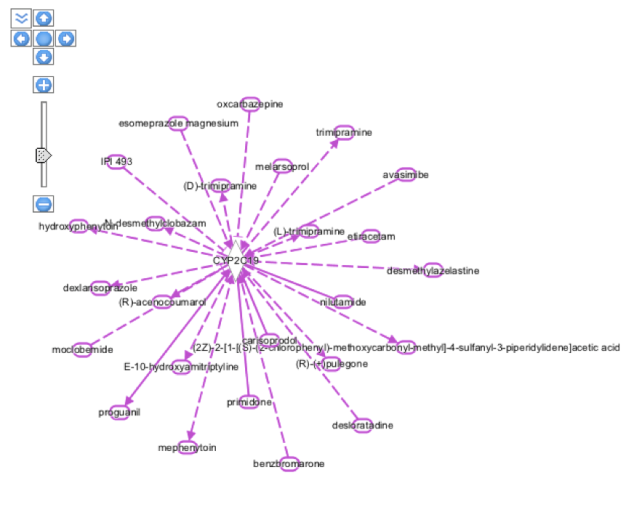
The pathway result in the above figure involves all molecular interactions for human,rat as well as mouse species. In the next step, some of the filtering options were selected to get a more precise view of the interactions of the CYP2C19 gene with other enzymes, molecules,chemicals ad drugs. For this, the MicroRNA-mRNA interactions from the Data Sources was deselected and along with that Moderated(Predicted) Confidence level was excluded, the species search was narrowed down to Human and Mouse and microRNA was excluded from the Molecule Types. After applying all these filters we were able to get the following view:

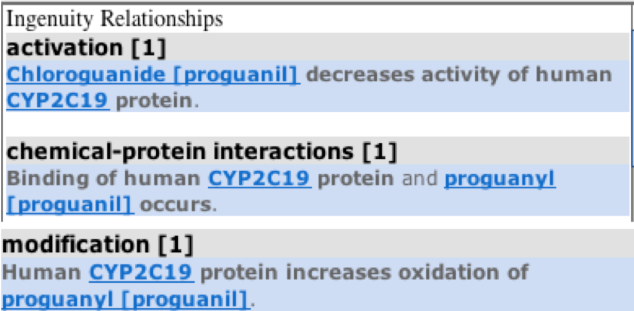
Hovering on individual molecules, gives information about the family to which they belong as highlighted in orange boxes in the diagram. For example, mephenytoin is a chemical drug whereas FOXA3 is a Transcription-regulator. Also, if we click on the relationship interaction solid or dotted line between the CYP2C19 gene and the molecule ( e.g blue colored line seen between chemical drug proguanil and the CYP2C19 gene), than it gives the summary of the relationship between the gene and the molecule as below:
Another feature to navigate through in IPA is the Diseases and Functions tool in which one can enter the disease name with which a particular gene is related and than add it to the existing pathway of molecules to see the relationship summary as below:

Based on my previous blog posts, we have the knowledge that the presence of a CYP2C19 gene allele in an individual leads to a poor metabolism of drugs like warfarin, mephenytoin,omeprazole, proguanil,etc. So , I selected he responsiveness of blood platelets disease from the various diseases list that was generated with CYP2C19 gene as entry and added it to my pathway. On clicking over the relationship arrow, the relationship summary was generated as seen in the above figure which is consistent with our knowledge from the past regarding poor metabolizing effect in mutant human CYP2C19 protein carriers.
Next,using the Trim Tool we can view the pathway with the specification such as show only those interactions which are direct between the gene and the molecules.We can also specify which species to be included, relationship types,molecule types, tissues and cell lines, diseases,biomarkers,etc. For, the CYP2C19 gene , the pathway was trimmed to show only direct interactions ( the species were not specified and thus included Human,Mouse ,Rat and Uncategorized species):

From approximately 25 interactions that were seen in previous images, trimming to direct interactions yields around 7-8 interactions only as shown in the above diagram.
“Canonical Pathways,” are idealized or generalized pathways that represent common properties of a particular signaling module or pathway. To view the involvement of a the CYP2C19 gene in a particular signaling pathway, the overlay button from Path Editor window was used and than Canonical pathway option was selected from the tools. This gave us a list of signaling pathways of which CYP2C19 gene is a part.The PXR/RXR signaling pathway was selected for analysis purpose as represented below:

The position highlighted in red is where the role of the CYP2C19 gene is in metabolism of various drugs.The Pathway and Tox lists feature of IPA also generates a report for the selected Canonical pathway. As per the summary provided by this report,The pregnane X receptor (PXR) is a nuclear receptor which is mainly expressed in the liver and intestine and along with the retinoid X receptor (RXR), it plays a role in metabolism of various drugs by activating the cytochrome P450 family of enzymes. Also,PXR/RXR activation induces drug conjugation enzymes as well as the drug efflux pumps. Thus, this PXR/RXR canonical pathway is very important for drug metabolism and transport and is also involved in bile acid synthesis and lipid metabolism. It also regulates the expression of gene products required for xenobiotic and endobiotic metabolism. This summary is in consonance with the information in my previous Gene Expression Analysis blog as well as from OMIM, which states that the CYP2C19 gene belongs to the cytochrome P450 family and is expressed mainly in liver and has a major role in metabolism of drugs such as mephenytoin, omeprazole,etc. Also, it is now known that individuals who are carrier of mutant CYP2C19 gene alleles are poor metabolizers of these drugs and from this signaling pathway we can explain the same.